
Bioinformatics is a huge part of modern biological study. In this series of posts, I’m going to introduce you to some of the bioinformatics tools and techniques that field biologists, such as myself, use in our daily work.

What you’ll learn
- The theory behind BLAST, one of the most widely-deployed bioinformatic algorithms.
- How to install BLAST on a fresh Ubuntu 16.04 LTS instance.
- How to perform a query against a precomputed database of sequences.
Prerequisites
To follow this guide, you’ll need:
- to know how to create a new Exoscale instance and how to to connect to it
- to be comfortable working in a Linux terminal
- a medium (4GB RAM or bigger) Exoscale instance running Ubuntu 16.04 LTS.
Sequence homology
First, let’s cover some background.
Within biology, two genes are defined as homologues if they come from a common source of ancestry; i.e. a particular organism. When species start to diverge from each other, due to migration, a natural event or an evolutionary constraint, their gene pools tend to accumulate different mutations over time.
One might expect that genes between two species that are closely related would look more similar than genes from species that had deviated from each other a long time ago. That usually holds true and this is the basis for most evolutionary and phylogenetic analyses.
Today, thousands of genomes from different species have been sequenced and are available for us to work with, revealing valuable information about their genes and their corresponding function. This proves particularly useful when researches get ahold of newly sequenced data, as it is important to be able to check if it looks similar to other already known sequences.
Because the current corpus of known data is extremely big, we need specialised tools to search it efficiently and to measure the degree of similarity between sets of results. That’s where tools like BLAST come into play.
What is BLAST and why is it useful?
BLAST is a family of algorithms designed for retrieving sets of data, similar to query strings, from a significantly large body of data. Unlike the algorithms that came before, BLAST uses a heuristic approach. While it doesn’t promise always to yield an optimal result, it can be orders of magnitude faster than the previous alternatives.
The BLAST algorithms were first published by Altschul et al. (1990). I encourage you to check out the reference for yourself, but in the meantime let’s take a quick look at how it works and what makes it so fast.
Before BLAST, an exhaustive comparison between two sequences would take a relatively long time to perform. If you scale this problem to thousands of sequences you will soon find that, regardless of the computational power you put into it, it will quickly become unfeasible. Compared with those older approaches, BLAST takes a couple of steps to significantly reduce the complexity of the problem:
- Your input sequence is analyzed and low-complexity regions (think of repeating patterns) that do not add meaningful information to the search are stripped.
- The sequence is broken down into words of a fixed length (usually three aminoacids or eleven DNA bases). These words are compared against a precomputed index of known words to find quickly an initial set of target sequences that could become a good match against your input. These initial pairs of matching words are called seeds.
- The seeds are then scored according to a set of rules that takes into account the probability of a particular sequence to appear in nature. BLAST will then try to extend these seeds into longer sequences, giving preference to the ones that tend to grow toward a higher score.
- When a resulting seed goes beyond a particular user-defined threshold it is reported as a result. BLAST then performs an exhaustive pairwise alignment over these gathered results and reports this data back to the user.
The speed at which BLAST arrives at its results allowed a new era of comparative bioinformatics to thrive, and nowadays, most genes get their function inferred by tools that automatically look for other genes with a high sequence similarity.
Downloading and installing BLAST
BLAST’s code is open source and freely available from the USA’s National Centre for Biotechnology Information.
You can find the source code and precompiled package at the following FTP site:
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
To determine the particular package to download, you need to know the architecture of the current instance you are using. All of Exoscale’s Linux instances are built over a 64-bit architecture, but just in case, here’s how you would check your own with a quick linux command:
uname -i
If you get back x86_64 then you are running in a 64-bit architecture.
To list the files available at the FTP site we will make use of a tool called lftp. You can install it quickly by issuing:
sudo apt-get install lftp
After that, the following command will print out the files available at /blast/executables/blast+/LATEST/ on the ftp.ncbi.nlm.nih.gov site:
lftp -e "cd blast/executables/LATEST; dir; quit" ftp.ncbi.nlm.nih.gov | awk '{print $NF}'
You should get back something like:
ncbi-blast-2.3.0+-1.src.rpm
ncbi-blast-2.3.0+-1.x86_64.rpm
ncbi-blast-2.3.0+-src.tar.gz
ncbi-blast-2.3.0+-src.zip
ncbi-blast-2.3.0+-universal-macosx.tar.gz
ncbi-blast-2.3.0+-win64.exe
ncbi-blast-2.3.0+-x64-linux.tar.gz
ncbi-blast-2.3.0+-x64-win64.tar.gz
ncbi-blast-2.3.0+.dmg
...
Choose the one that’s appropriate for you, in this case ncbi-blast-2.3.0+-x64-linux.tar.gz, and download it using wget:
wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/ncbi-blast-2.3.0+-x64-linux.tar.gz
When you’re done, you should see something like this:
Decompress this file and cd into the resulting directory to find a precompiled distribution of BLAST.
tar -xzvf ncbi-blast-2.3.0+-x64-linux.tar.gz
# Optional, remove the compressed package you downloaded
rm ncbi-blast-2.3.0+-x64-linux.tar.gz
cd ncbi-blast-2.3.0+
Inside, you’ll find a bin directory. This is where the BLAST executables live. Here’s what a few of them do:
- blastdbcmd: Create and managed BLAST databases.
- blastn: Compare DNA sequences against a database of DNA sequences.
- blastp: Compare protein sequences against a database of protein sequences.
The others are helpful when we need to translate between different sequence domains (DNA/Protein; blastx, tblastn, tblastx) or when we need to perform an analysis following a different search and scoring strategy (psiblast, rpsblast). Don’t worry about them for now, as we will cover them in later editions of these tutorials.
To set up BLAST, we need to add these executables to our environment’s PATH variable. The easiest way to do this is to enter the bin directory and then run:
export PATH=$PATH:$PWD
You probably already know that $PWD is an environment variable that holds the location of our current working directory. After running this command you should be able to execute any of the BLAST programs from your current shell. If you want to make this available permanently, you’d need to add the following line to your $HOME/.bash_profile file.
export PATH=$PATH:<PATH_TO_YOUR_BLAST_BIN_DIRECTORY>
If you don’t have a $HOME/.bash_profile file, just create one. Recall that $HOME refers to the location of your home directory.
After you’ve done this, you can check that BLAST is working correctly by running:
blastn -version
If all goes fine, there should be no errors and you should get back something like this:
blastn: 2.3.0+
Package: blast 2.3.0, build Nov 30 2015 13:03:37
The final step for setting up BLAST is to create a directory to store the sequence databases that you’ll be using. The BLAST documentation suggests you do this inside your BLAST folder but I’d recommend that you do this in another folder outside this one. Why? Because that way, if you download a newer version of BLAST in a different location you could just delete your previous one without worriying that your databases are stored there.
Let’s assume that you chose to store your databases at $HOME/blast_db. You just need to define a BLASTDB variable with the location of this directory.
mkdir $HOME/blast_db
export BLASTDB=$HOME/blast_db
And remember to add this last line, too, to your $HOME/.bash_profile.
Downloading a precomputed sequence database from NCBI
On the same FTP site where you just downloaded BLAST, you can find many precomputed databases that may be useful for you depending on your particular use case. This data is stored under the /blast/db/ directory. You can find a file at /blast/documents/blastdb.html with information about the data that makes up each of these files.
refseq_rna.00.tar.gz through ferseq_rna.08.tar.gz contain all RNA sequences derived from all publicly available transcriptomic records. For the sake of this tutorial, we will download only the first file, refseq_rna.00.tar.gz, but be aware that when you actually do some work with this full set you would have to download all of these files. Change your location to your BLASTDB directory and download this file with:
cd $BLASTDB
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/refseq_rna.00.tar.gz
When finished, you should see something similar to when you downloaded the BLAST suite.
Although it is compressed, refseq_rna.00.tar.gz still measures just shy of 1GB. You will find out that, in order to work with genomic data, you need to have a lot of disk space at hand and a moderately fast internet connection if you don’t want to wait for days when downloading it. One of the advantages of performing this analysis on an Exoscale instance is that you already have as much disk space as you’d need to and a solid network infrastructure at your disposal allowing you to download all this data much faster. All of this, while keeping your information secure and private, because you are the only one who can access your data or control who else does.
Uncompress this file and remove it with the following commands:
tar -xzvf refseq_rna.00.tar.gz
rm refseq_rna.00.tar.gz
Your BLASTDB directory should now have the following files:
refseq_rna.00.nhr refseq_rna.00.nnd refseq_rna.00.nog refseq_rna.00.nsi refseq_rna.nal
refseq_rna.00.nin refseq_rna.00.nni refseq_rna.00.nsd refseq_rna.00.nsq
Those files are the indexes and metadata needed for BLAST to perform queries against. We are now ready to start searching among this data with any of the BLAST executables.
Performing a BLAST query against a precomputed database
In order to perform a BLAST search, you need to provide a FASTA file with the input sequence (or sequences) that you want to find homologues of. A FASTA file is a regular text file with a specific, but simple, format that looks like this:
>gi|4557562|ref|NM_000122.1| Homo sapiens ERCC excision repair 3, TFIIH core complex helicase subunit (ERCC3), transcript variant 1, mRNA
GGGAGCTTCCGGATTGAGCCGGAAGTCCCCCCAGAGCGGATGCCGCGGCGGGCCTGTGGGAGCGGGGTCATCTTCTCTCTGCTGCTGTAGCTGCCATGGGCAAAAGAGACCGAGCGGACCGCGACAAGAAGAAATCCAGGAAGCGGCACTATGAGGATGAAGAGGATGATGAAGAGGACGCCCCGGGGAACGACCCTCAGGAAGCGGTTCCCTCGGCGGCGGGGAAGCAGGTGGATGAGTCAGGCACCAAAGTGGATGAATATGGAGCCAAGGACTACAGGCTGCAAATGCCGCTGAAGGACGACCACACCTCCAGGCCC
As you can see, a FASTA file is a collection of sequences where each of them is preceded by a header, identified by a line that starts with > and which contains the metadata associated with the sequence that follows.
Create a new text file called example.fa and copy/paste the example shown previously. You can use any input file you may have and it can have any filename as well.
example.fa contains a DNA sequence and we are going to search against a database of DNA sequences, so we are going to use the program blastn. We will execute blastn with the following parameters:
- -query : The name of the file to be used as input.
- -db : The name of the database we want to query against to.
-
-out : The name of the file where BLAST will write out the results. If we do not specify this, it will print out the results to standard output.
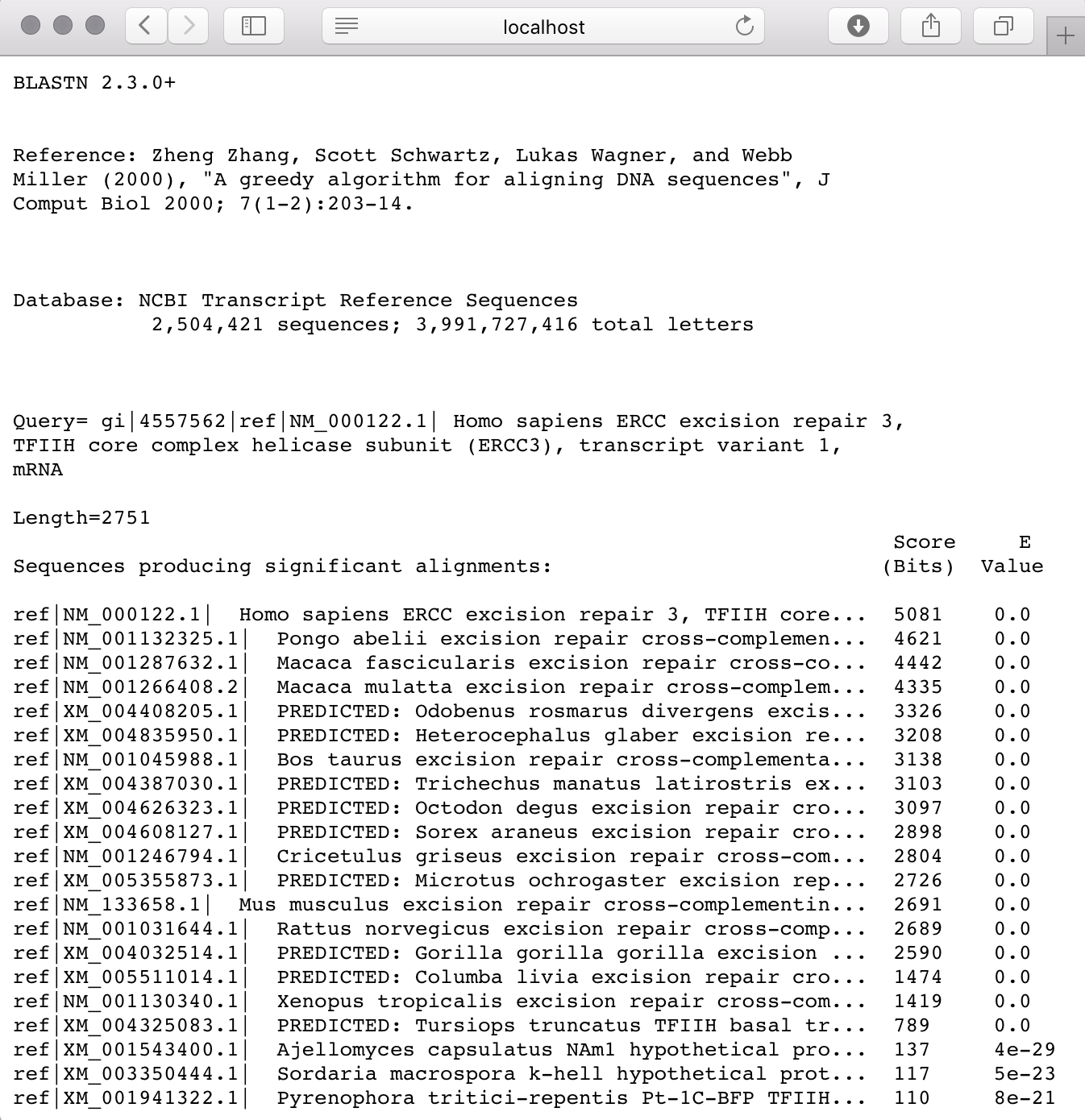
blastn -query example.fa -db refseq_rna.00 -out results.txt
Now, you should have a results.txt file with the results of this query. Take a look at it using cat or your preferred text editor.
A small bonus: viewing your results using your web browser
Working with these files becomes cumbersome because their length easily exceeds the viewport of your terminal.
I wont go into detail about how any of this works as that would escape the purpose of this BLAST tutorial, but I will show you, very quickly, how you can set up an http server and make these files available over the web.
Don’t worry! You’ll be the only one who can see them.
Download and install nodejs and npm on your Exoscale instance:
sudo apt-get install nodejs-legacy npm
Verify that they were installed correctly:
node -v
npm -v
You should get something like:
v4.2.6
3.5.2
Install http-server:
npm install -g http-server
Now, go to the location where the files you want to see are stored (or to your $HOME directory), and execute:
http-server
You should see something like:
Starting up http-server, serving ./
Available on:
http://127.0.0.1:8080
http://<YOUR_IP>:8080
Hit CTRL-C to stop the server
You just set up a web server on your instance, listening to requests at port 8080. This port is not open in your instance by default, so it is not accesible by the public. Instead, we are going to route it to your local computer with an SSH tunnel.
Open up a new terminal on your computer (note: not your Exoscale instance) and execute:
ssh -NL 8080:localhost:8080 <YOUR_INSTANCE_USER>@<YOUR_INSTANCE_IP>
If you want to know how all of this works, you should read up about SSH Tunnels. But for now, you just forwarded port 8080 on Exoscale’s instance to your local computer, so you can open any web browser, navigate to http://localhost:8080/ and voila, see your files in there.
Navigate to your results.txt file and you should see your work displayed in a much more user-friendly environment.
That’s it for now, stay tuned for the second part, where we’ll show you how to set up your own private BLAST databases and start submitting queries against them.


